抗がん剤で心筋が萎縮する機序を解明
~抗がん剤の副作用軽減に期待~
- 発表者
- 島内 司 (九州大学病院 手術部 助教)
冨田 拓郎 (自然科学研究機構 生理学研究所心循環シグナル研究部門 助教)
伊藤 智哉 (クィーンメアリー大学医歯学部ウィリアムハーヴェイ研究所 リサーチフェロー)
西村 明幸 (自然科学研究機構 生理学研究所心循環シグナル研究部門 特任助教)
松金 良祐 (国立がん研究センター基盤的臨床開発研究コアセンター 特任研究補助員)
小田 紗矢香(総合研究大学院 大学5年一貫 博士課程2年)
外 須美夫 (九州大学医学研究院 臨床医学部門 外科学講座 教授)
井手 友美 (九州大学医学研究院 附属心臓血管研究施設 講師)
小板橋 紀通(群馬大学大学院医学系研究科内科学講座 循環器内科学 助教)
内田 浩二 (東京大学大学院農学生命科学研究科 応用生命化学専攻 教授)
森 泰生 (京都大学大学院工学研究科 合成・生物化学専攻 教授)
西田 基宏 (自然科学研究機構 生理学研究所心循環シグナル研究部門 教授
九州大学 大学院薬学研究院 創薬育薬研究施設統括室 教授(併任) )
発表概要
抗がん剤を用いる化学療法は、全身性がん治療の第一選択です。しかし、抗がん剤は疲労感/倦怠感や筋肉痛、ひどい場合は心筋症といった副作用を起こすことが問題視されています。原因は心筋や骨格筋などの萎縮であることは知られていましたが、抗がん剤が筋萎縮を起こす機構は不明でした。
今回、生理研、九州大学、群馬大学、東京大学、京都大学との共同研究において、心筋細胞膜に存在し、抗がん剤投与により発現増加するTRPC3チャネルが、活性酸素を発生することで心筋細胞を萎縮することを発見。実際、TRPC3チャネルを阻害する化合物が、抗がん剤誘発性の心不全を軽減することを明らかにしました。
発表内容
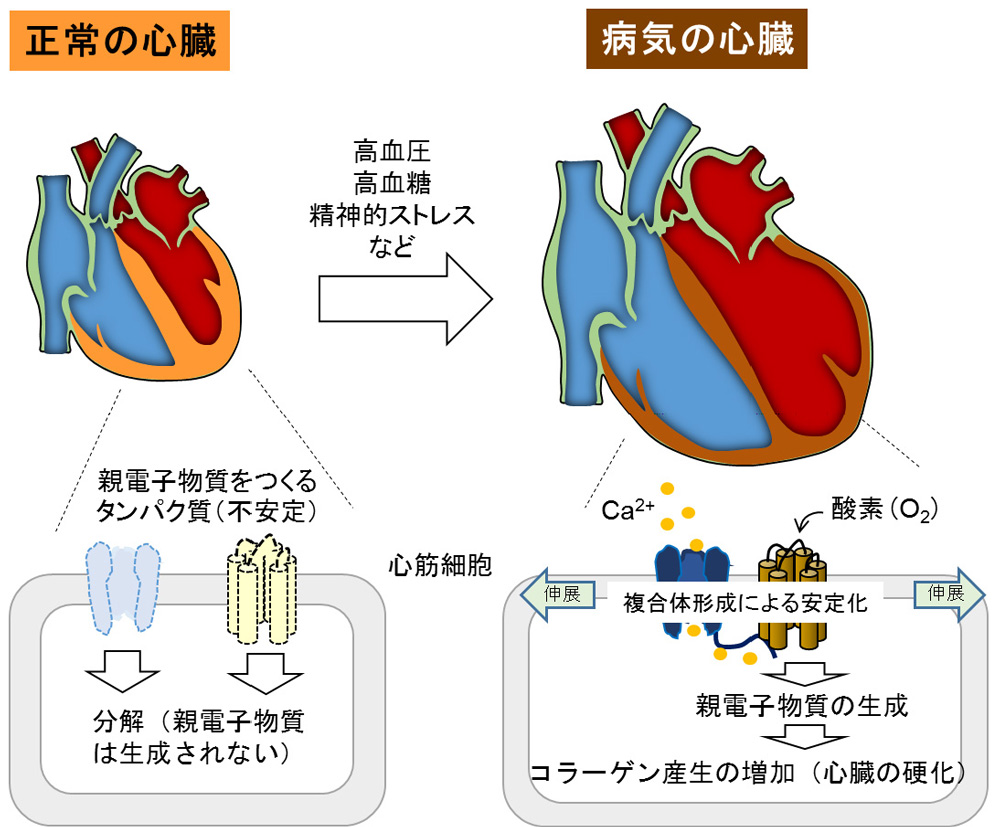
図1 (昨年度の研究成果)圧負荷心臓におけるTRPC3-Nox2タンパク質複合体形成を介したコラーゲン産生増加(心臓の硬化)
(拡大画像↗)
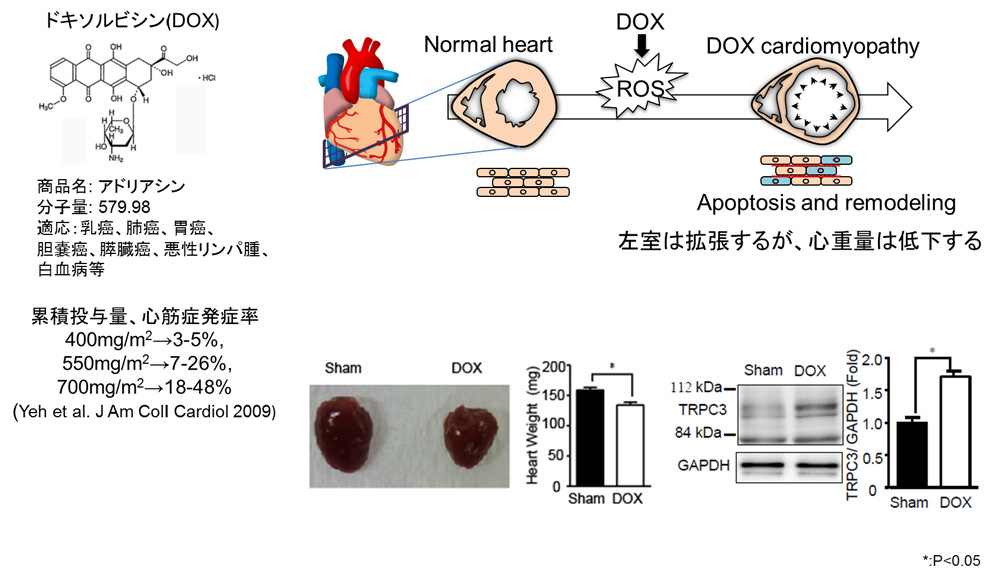
図2 TRPC3-Nox2複合体形成はドキソルビシン(DOX)誘発性の心筋萎縮(心不全)も仲介する。
(拡大画像↗)
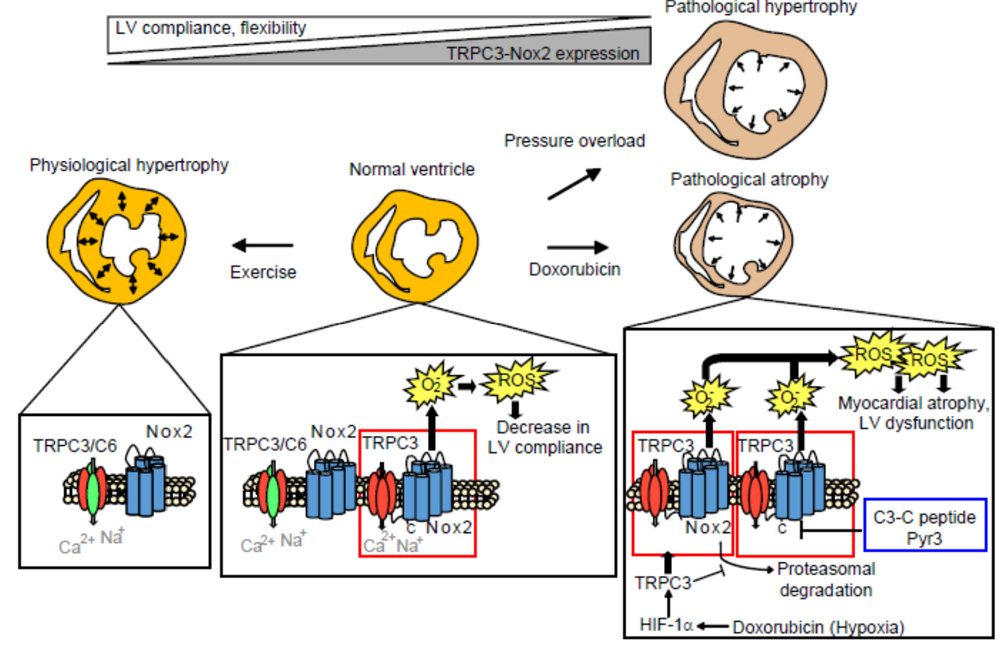
図3 TRPC3チャネル阻害は自発的な運動と同様に、ドキソルビシン誘発性心筋萎縮を抑制する(TRPC3阻害は運動を模倣する?)
(拡大画像↗)
がんは、我が国で最も死亡率の高い病気であり、日本人の2人に1人が罹患するといわれています。抗がん剤治療(化学療法)は、手術および放射線治療と並ぶ「がん治療の3本柱」の1つとして、高い医療実績を誇っています。その一方で、化学療法を続けることにより、筋力低下による疲労感/倦怠感や廃用(寝たきり)症候群、心筋症といった副作用が起こりうることが問題視されています。しかし、抗がん剤が筋力低下を起こす原因についてはよくわかっていませんでした。
私たちのグループは昨年、心筋細胞膜上に存在するCa2+透過型カチオンチャネル(transient receptor potential canonical (TRPC) 3)が、酸化ストレスの原因となる活性酸素の生成酵素である細胞膜タンパク質NADPHオキシダーゼ2(Nox2)と相互作用し、Nox2タンパク質の分解を抑制(安定化に寄与)していることを報告しました。さらにTRPC3チャネルは心筋細胞の物理的伸展刺激により活性化し、Nox2からの活性酸素生成を促すことで、心臓の線維化(硬化)を誘導することも明らかにしてきました(図1)。
今回私たちは、高用量のアントラサイクリン系抗がん剤ドキソルビシン(商品名:アドリアシン)が、心臓において急性期にTRPC3-Nox2タンパク複合体数を増加し、酸化ストレスを誘発することで心筋細胞を萎縮させることを、マウスを用いて明らかにしました(図2)。ドキソルビシン投与は、心筋細胞の「低酸素化」を誘発することでTRPC3タンパク質の発現を増加し、TRPC3がNox2タンパク質を安定化することでNox2依存的な活性酸素の生成を促し、結果的に心筋細胞を萎縮させることがわかりました。TRPC3とNox2の相互作用を特異的に阻害するタンパク質をマウスの心筋細胞特異的に発現させたところ、ドキソルビシン投与によるマウスの心筋萎縮と心機能低下が軽減されました。さらに、TRPC3チャネルを阻害することが報告されている複数の化合物の中から、TRPC3-Nox2複合体形成も抑制できる化合物pyrazole-3を同定し、pyrazole-3がドキソルビシン誘発性の心筋萎縮を顕著に抑制することも明らかにしました。
一方、適度な運動がマウスのドキソルビシン心毒性を軽減することも過去に報告されています。適度な運動を与え続けたアスリートモデルマウスの心臓ではTRPC3-Nox2複合体形成が抑制されていました。TRPC3遺伝子欠損マウスの心機能を解析した結果、やはりアスリートモデルマウスの心臓と同様にコンプライアンス(弾性と伸展性)の高い柔軟な心筋を呈していることがわかりました。TRPC3チャネル活性だけを変化させても心筋の柔軟性は変化しないことから、TRPC3-Nox2複合体形成の阻害が、適度な運動を模倣することで、抗がん剤投与による急性期の心筋萎縮を抑制する可能性が示されました(図3)。
本研究により、TRPC3-Nox2複合体形成が抗がん剤誘発性の心筋萎縮を引き起こす原因となることが明らかとなりました。今後、TRPC3チャネル活性およびTRPC3-Nox2相互作用を阻害する薬と抗がん剤の併用療法の開発が、健康長寿社会の実現や医療経済的負担の軽減に大きく貢献するものと期待されます。
本研究は国立研究開発法人科学技術振興機構(JST)・戦略的創造研究推進事業(さきがけ)の研究領域「疾患における代謝産物の解析および代謝制御に基づく革新的医療基盤技術の創出」における研究開発課題「硫黄循環・代謝を基軸とした生体レドックス恒常性制御基盤の構築」(代表:西田 基宏教授)の一環で行われたと共に、日本学術振興会の科学研究費補助金、文部科学省の新学術領域「酸素生物学」、内藤記念財団、大幸財団などの研究助成、ならびに日本医療研究開発機構(AMED)「創薬等ライフサイエンス研究支援基盤事業(九大拠点)」による支援を受けて行われました。
< この研究の社会的意義>
<今回の発見>
2. ドキソルビシンは、心筋細胞の低酸素化を誘導することでTRPC3タンパク質の発現を増加し、Nox2を安定化することを明らかにしました。
3. TRPC3とNox2タンパク質の相互作用を仲介するタンパク質(TRPC3のNox2と結合する部分配列)を心筋細胞特異的に発現させたマウスで、ドキソルビシン誘発性の心筋萎縮が抑制されました。
4. 既存のTRPC3チャネル阻害化合物の中からTRPC3-Nox2複合体形成も抑制しうる化合物pyrazole-3を同定し、pyrazole-3がドキソルビシン誘発性の心筋萎縮と心機能低下を抑制することを明らかにしました。
5. 適度な運動を与え続けたアスリートモデルマウスでは、心筋TRPC3-Nox2複合体形成が顕著に抑制されていました。
6. TRPC3遺伝子欠損マウスの心機能を解析したところ、アスリートモデルマウスの心臓と同様、柔軟なコンプライアンス(弾性と伸展性)をもつ心筋を呈することがわかりました。
発表雑誌
- 雑誌名
- :Journal of ClinicaI Insight 2(15):e93358(2017)(掲載日:2017年8月3日)
- 論文タイトル
- :TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy
- 著者
- :Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N, Uchida K, Sumimoto H, Mori Y and Nishida M.
- DOI番号
- :10.1172/jci.insight.93358
- 論文URL
- :https://doi.org/10.1172/jci.insight.93358
問い合わせ先
-
<研究について>
東京大学大学院 農学生命科学研究科 応用生命化学専攻食糧化学研究室
教授 内田 浩二(ウチダ コウジ)
Tel:03-5841-5128
Fax:03-5841-8026
Email: a-uchida<アット>mail.ecc.u-tokyo.ac.jp <アット>を@に変えてください。
大学共同利用機関法人 自然科学研究機構 生理学研究所心循環シグナル研究部門
九州大学 大学院薬学研究院 創薬育薬研究施設統括室(併任)
教授 西田 基宏(ニシダ モトヒロ)
TEL: 0564-59-5560
Email: nishida<アット>nips.ac.jp <アット>を@に変えてください。